
近期,FDA在审评乌司奴单抗(Stelara)的生物类似药时,首次豁免了临床疗效研究。这是FDA历史上首次豁免单抗类生物类似药临床疗效试验,或将开启生物类似药审评的根本性变革。
值得一提的是,生物类似药III期临床豁免后,企业能显著缩短开发周期,节省约50%的研发成本。
但是,任何制度转折都伴随着新的问题和潜在风险,如果说过去的挑战是成本过高、周期过长,那么未来的挑战则集中在如何维持社会信任、科学严谨与市场秩序之间的平衡。
研发成本砍半
FDA豁免单抗生物类似药临床疗效研究,不仅是一项监管文件的调整,更是对整个产业格局的重塑。它首先作用于研发成本和周期,然后传导到市场竞争和价格体系,最终影响到不同国家和不同规模企业的战略选择。
对于中国制药企业来说,这既是一扇难得的窗口,也是一道新的考题。
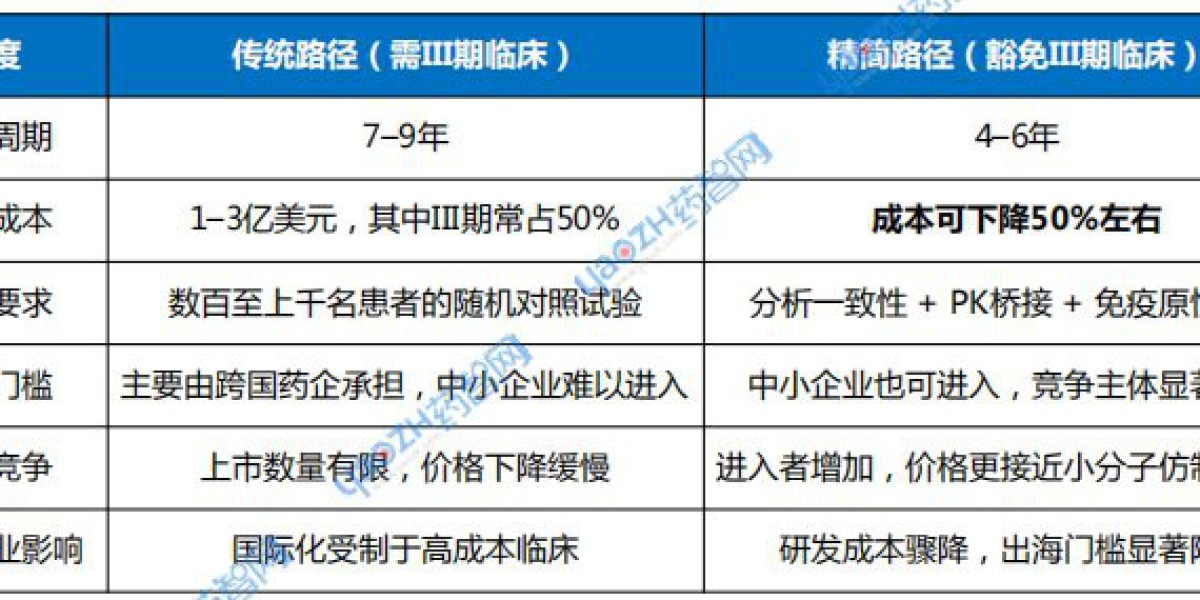
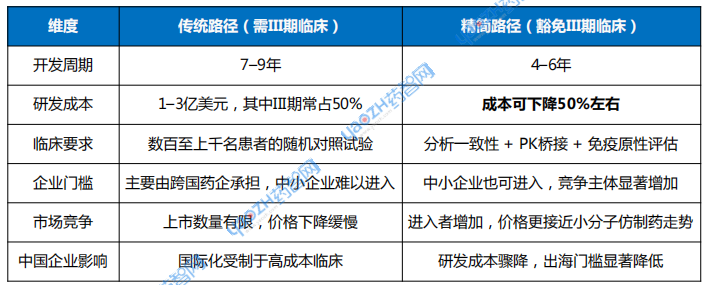
在传统路径下,开发一个单抗生物类似药往往需要七到九年,资金投入超过一亿至三亿美元,其中大规模的III期临床研究常常占到总预算的一半。很多企业在进入生物类似药开发时并非缺乏技术,而是被成本与周期劝退。豁免III期之后,开发周期有望缩短两到三年,研发费用下降50%,原本需要大规模现金流支撑的上市道路,突然变成了中型企业也能负担的项目。这一变化有望直接重塑行业格局。
表1 传统生物类似药与豁免III期临床生物类似药上市对比
数据来源:参考资料
对于跨国药企来说,这意味着竞争压力骤然增加。过去,大药企凭借雄厚资金与成熟临床体系,在生物类似药市场上形成天然垄断。如今,这道护城河被削弱,中小企业甚至初创公司都有机会进入。市场上参与者数量的增加,势必会带来更激烈的价格竞争。
这种趋势对中国企业尤为重要。过去几年,中国已逐渐成长为全球生物类似药的重要供应力量。齐鲁制药、信达生物、百奥泰、复宏汉霖、君实生物等一批企业在国内市场陆续推出了利妥昔单抗、阿达木单抗、贝伐珠单抗、曲妥珠单抗等多个品种的生物类似药,并在部分品种上实现了医保放量。它们面临的核心挑战是:如何突破国内价格竞争的红海,进入更高价值的欧美市场。在这一过程中,昂贵的III期临床试验要求一直是过去十年制约中国企业国际化的障碍之一。
随着FDA豁免政策落地,这一障碍有望迅速削弱。以往需要数千万美元投入的III期临床,如今有获得豁免的可能,企业只需把资源集中在分析一致性和工艺控制上。对于已经拥有完备生产体系和分析能力的中国企业来说,这是缩短与跨国药企差距的重要契机。它们可以用远低于跨国药企的成本,在相对短的时间内,在满足合规与商业化能力要求的前提下,将生物类似药推向美国和欧洲市场。这不仅意味着更高的利润空间,也可能使中国企业在全球药品供应链中占据更重要的位置。
当然,机会并不意味着无条件的成功。豁免III期后,监管的重心会前移到质量控制和分析一致性。这要求企业必须具备最先进的质谱、糖基化分析、功能性检测等平台,并且在工艺稳定性、批次间一致性和免疫原性研究上做到满足严格的监管要求。这对中国企业既是机遇,也是压力。
另外一点是时间窗口。Stelara生物类似药只是开始,未来还有Keytruda、Dupixent等一系列单抗将进入专利到期和生物类似药竞争期。谁能在最短时间内完成申报,谁就能抢占先发优势。过去的经验显示,首仿往往能占据40%甚至更高的市场份额,而后续进入者则面临更为有限的市场空间。对于中国企业而言,这意味着必须快速提升全球注册策略能力,建立跨国申报与商业化合作网络。
科学严谨与市场秩序的平衡
FDA豁免单抗生物类似药的III期临床要求,为行业带来了前所未有的便利,但任何制度转折都伴随着新的问题和潜在风险。如果说过去的挑战是成本过高、周期过长,那么未来的挑战则集中在如何维持社会信任、科学严谨与市场秩序之间的平衡。
首先是监管信任的问题。长期以来,医生和患者之所以逐步接受生物类似药,一个重要原因在于他们知道这些产品经过了与原研药类似的临床试验验证。如今,FDA在特定情况下不再强制要求这一环节,虽然在科学上有充分逻辑支撑,但在公众认知层面可能并不容易被立即接受。一些医生可能会产生疑虑:没有真实患者验证的生物类似药,是否足以替代原研药?患者也可能因此产生顾虑,影响药物的使用率和市场渗透。对监管机构而言,如何通过透明的解释、教育和后续数据发布来维持公众信心,将成为新时期的重点任务。
其次是免疫原性和罕见安全性事件的风险。III期临床虽然样本有限,但至少可以提前观察几百名患者的用药情况,而在豁免临床的情境下,上市前的数据主要来自小规模PK桥接与实验室免疫原性研究。这类研究对大部分常见风险已经足够敏感,但对于百万分之一的低频免疫事件,仍然可能难以捕捉。因此,风险并没有消失,而是被转移到了上市后的药物警戒环节。企业必须建立完善的风险管理计划(RMP),监管机构也必须强化真实世界数据的采集和分析,确保一旦出现信号能够快速响应。对于缺乏长期国际药物警戒经验的中国企业来说,这可能是进入欧美市场后面临的首要考验。
第三是支付方和市场策略的不确定性。理论上,研发成本的大幅下降应当推动生物类似药价格更接近小分子仿制药的走势,但这并不完全取决于制造商。美国市场的药价形成高度依赖PBM(药品福利管理机构)的谈判和回扣机制,欧洲则受到各国医保机构集中采购的强烈影响。在这种格局下,即便企业能够低价生产,也未必能立刻传导到患者终端。支付方在价格谈判中可能仍会维持一定的利润空间,导致降价幅度和速度不如预期。对中国企业而言,如何理解和驾驭支付体系,是能否真正从低成本优势转化为市场份额的关键。
最后是不确定的国际化挑战。FDA和EMA虽然在监管逻辑上趋同,但在具体执行上仍然可能存在差异。例如,是否所有单抗都能豁免III期,还是需要逐案评估?是否会对某些新机制或免疫风险较高的品种设置额外要求?这些问题在未来几年都会逐步显现。此外,即便在科学上被豁免,商业上的阻力依然存在。跨国药企可能会通过专利延伸、市场营销和医生教育来延缓生物类似药的渗透。对于中国企业而言,仅仅依靠低成本切入并不足以长期立足,更需要在质量、供应链稳定性和国际合作网络上建立声誉。
综上,FDA的豁免政策虽然降低了研发门槛,但也让行业面临新的试炼。监管信任、免疫原性风险、支付方策略和国际市场的不确定性,将决定未来生物类似药能否真正实现像小分子仿制药一样的普及。对于中国企业来说,这既是通往全球市场的钥匙,也是一次对综合能力的全面检验。
从监管松绑到产业重塑
FDA首次豁免单克隆抗体生物类似药临床疗效研究,标志着生物医药行业进入了一个新的阶段。这不仅仅是对乌司奴单抗生物类似药的一次个案处理,而是代表着监管逻辑从“重复验证”走向“科学合理性”的代际转换。
这一政策将深刻改变产业的运行方式。研发成本和周期大幅下降,更多的中小企业有机会进入竞争,价格曲线有望更快地逼近小分子仿制药的水平,全球患者将因此享受到更可负担的生物药治疗。与此同时,EMA、MHRA、加拿大等监管机构已经或正在走向相同的方向,国际监管的趋同让一次开发、多地申报成为现实,为全球生物类似药市场的活跃奠定了基础。
然而,新的机遇伴随着新的挑战。如何维护医生和患者的信任,如何在缺少III期临床“缓冲”的情况下通过真实世界数据和风险管理来保证安全性,如何与支付方博弈以实现价格优势真正惠及患者,这些都将是生物类似药产业必须面对的考题。尤其对中国企业而言,监管大门的打开既是机会,也是考验。只有在质量体系、分析能力、工艺一致性和国际合作网络上建立过硬实力,才能真正把成本优势转化为市场份额。
回望小分子仿制药的历史,当年《哈奇–瓦克斯曼法案》引爆了美国仿制药产业的黄金时代,造就了一批全球性企业。今天,生物类似药或许正站在类似的转折点上。对于中国企业而言,这不仅是一次进入欧美市场的窗口,更可能是一次建立全球竞争力的契机。未来几年,谁能率先适应这一监管范式的转变,谁就有机会在新一轮的生物类似药竞争中脱颖而出。
Ref.
1.Fontanillo,M.et al.Three imperatives for R&D in biosimilars.McKinsey&Company.19.08.2022.
2.Moore,T.J.et al.Assessment of Availability,Clinical Testing,and US Food and Drug Administration Review of Biosimilar Biologic Products.JAMA Intern Med.2021 Jan 1;181(1):52-60.doi:10.1001/jamainternmed.2020.3997.PMID:33031559;PMCID:PMC7536628.
3.Streamlining the Development of Biosimilar Medicines.Biosimilars Council.31.05.2024.
4.Tuszyner,M.The New Era of Biosimilar Development:Seizing the Opportunity Under EMA’s Streamlined Guidelines.Mabion.Retrieved 08.09.2025.
5.Tu,S.S.et al.Accelerating biosimilar market access:the case for allowing earlier standing.J Law Biosci.2025 Jan 3;12(1):lsae030.doi:10.1093/jlb/lsae030.PMID:39759260;PMCID:PMC11697977.
6.McGovern,G.et al.FDA to Waive Clinical Efficacy Studies for Monoclonal Antibody Biosimilars.Pharmacy Times.02.09.2025.
声明:本内容仅用作医药行业信息传播,为作者独立观点,不代表药智网立场。如需转载,请务必注明文章作者和来源。













